 期刊:Science China-Life Sciences
期刊:Science China-Life Sciences
影响因子:8.0
成簇的规则间隔短回文重复序列 (CRISPR)/Cas9 系统是编辑哺乳动物基因组的革命性方法。随着慢病毒递送方法的发展,CRISPR 筛选技术出现,并实现了全基因组敲除的低成本方式。然而,CRISPR 筛选只能分析具有非常不同表型的基因,例如那些显着影响细胞生长的基因,或者那些可以用抗体或荧光蛋白直接检测到的基因。
2016 年,开发了一种称为单细胞 CRISPR 筛选 (scCRISPR-seq) 的新技术,该技术将 CRISPR 扰动和单细胞测序相结合,以实现大规模单细胞分辨率的混合遗传筛选。scCRISPR-seq 的关键技术创新是慢病毒载体的创造性设计,称为 Perturb-seq 载体,允许从测序中鉴定每个细胞中的 sgRNA(图 16A)。scCRISPR-seq 可以促进复杂调控机制和异质细胞群的高通量功能解剖。
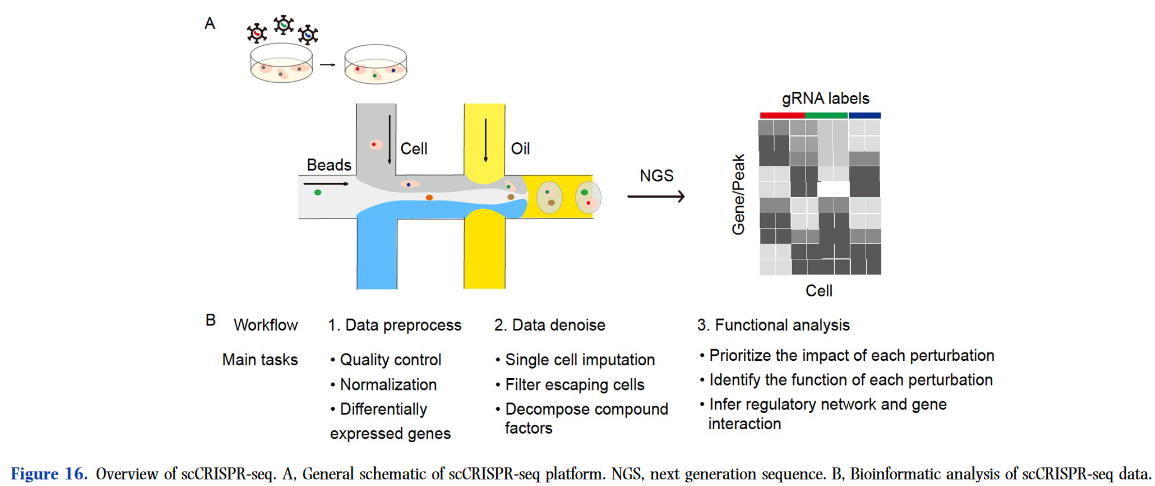
在本章中,我们将分四个不同的部分全面回顾 scCRISPR-seq。首先,我们将介绍 scCRISPR-seq 每个类别中的代表性技术。其次,我们将深入研究专门为分析 scCRISPR-seq 数据开发的主要工具。第三,我们将探索 scCRISPR-seq 的重要应用。最后,我们将得出结论并参与与 scCRISPR-seq 相关的局限性和未来趋势的讨论。
scCRISPR-seq 平台类别
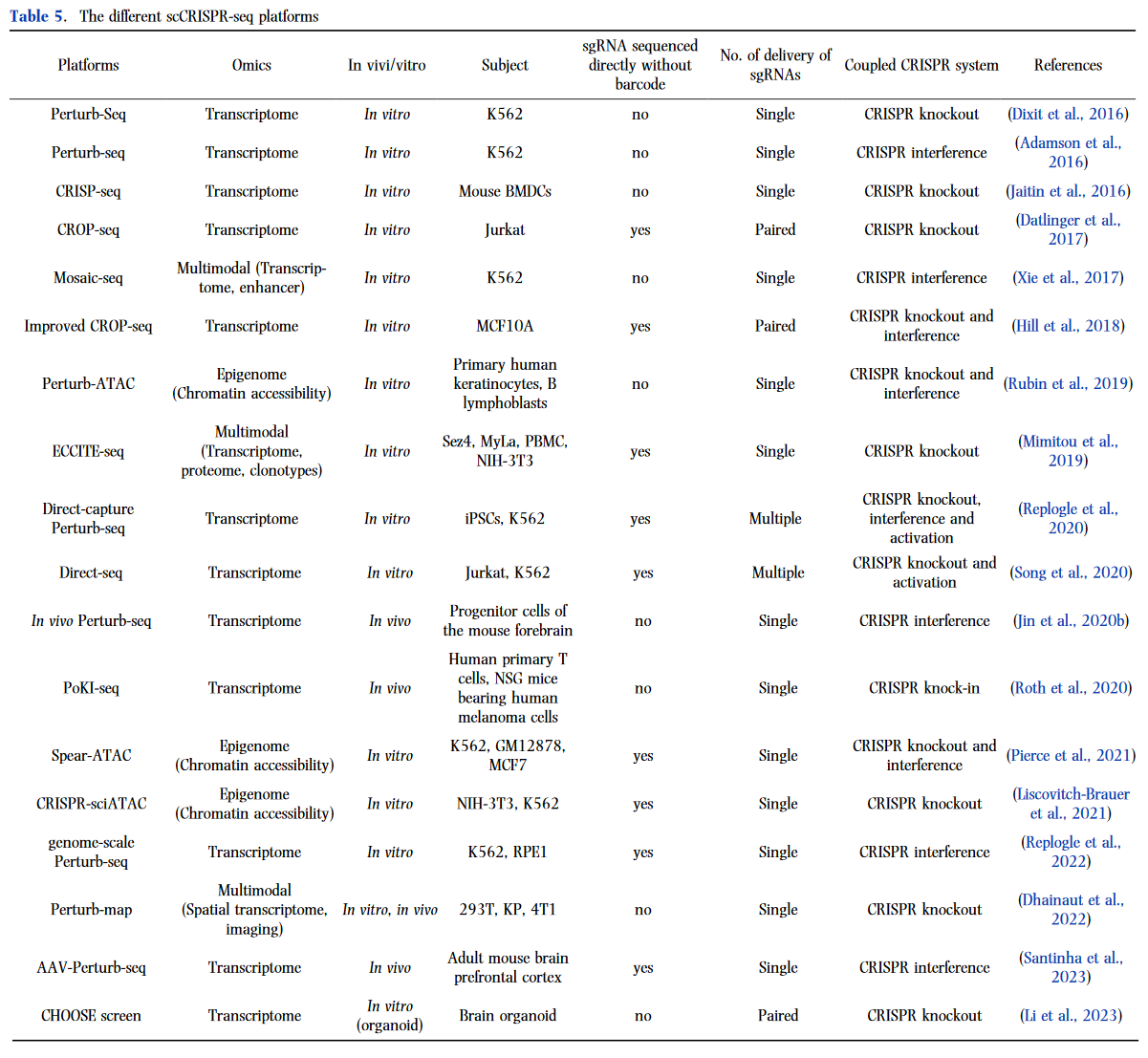
目前,已经出现了许多替代的 scCRISPR-seq 平台(表 5)。基于 scCRISPR-seq 的集成组学方法,这些平台可分为三大类:基于转录组的 scCRISPR-seq、基于表观基因组的 scCRISPR-seq 和多模态 scCRISPR-seq。
基于转录组的 scCRISPR-seq
主要的 scCRISPR-seq 平台是基于转录组的应用程序,将 CRISPR 筛选与单细胞 RNA-seq 相结合。对于基于转录组的 scCRISPR-seq,Perturbseq 载体通常由单向导 RNA (sgRNA)、细胞条形码 (CBC)、基因条形码 (GBC) 和 UMI 组成,例如 Perturb-seq和 CRISPseq。在 Perturb-seq 载体中,sgRNA 用于指导 Cas9 核酸酶在目标基因组区域诱导双链断裂,CBC 用于标记每个细胞,而 GBC 用于标记每个 sgRNA,UMI 用于标记每个转录本。Perutrb-seq 和 CRISP-seq 是第一个开发的 scCRISPR-seq 平台。这些方法涉及 Perturb-seq 载体的复杂构建,包括复杂的克隆策略,有时还需要 gRNA 间隔区与其条形码的解耦,这限制了它们的多功能性。CROPseq 优化了 Perturbseq 载体的设计,通过向 Perturb-seq 载体添加 Poly-A 尾部来检测每个细胞中诱导的 sgRNA 与 mRNA 偶联,这大大降低了 scCRISPR-seq 的复杂性和成本。然而,Hill 等人。(2018) 表明,由于这些研究的 Perturb-seq 载体设计,现有研究的慢病毒交换率仅为 50% 左右。因此,他们通过将向导 RNA 作为条形码来优化 CROP-seq 载体设计,将交换率提高到 94%。由于 Perturb-seq 载体设计的限制,Perturb-seq 和 CRISP-seq 的每个慢病毒载体只能向细胞递送单个编码的 sgRNA,而 CROP-seq 能够将成对的 sgRNA 递送到细胞。也就是说,它们都与多个 sgRNA 的递送不相容。为了解决这个问题,Replogle 等人。(2020) 设计了直接捕获 Perturb-seq,其中表达的 sgRNA 与单细胞转录组一起测序,并能够递送多个 sgRNA。直接捕获 Perturbseq 对于遗传相互作用的机制解剖特别有价值。它进一步降低了 Perturb-seq 实验的成本。Direct-seq 具有与直接捕获 Perturb-seq 类似的功能,可实现 CRISPR 扰动及其转录读数一起分析,并支持多个 sgRNA 的递送。2022 年,Replogle 等人引入了基因组规模的 Perturb-seq 方法。(2022 年),能够对影响 9,867 个基因的基因组规模遗传扰动进行公正和全面的分析。这一突破促进了系统的基因功能分配和复杂细胞表型的探索。最近,利塔尔.(2023) 开发了 CRISPR-人类类器官-单细胞 RNA 测序 (CHOOSE) 系统。这种创新系统能够进行遗传破坏和单细胞转录组学,用于混合功能丧失筛选嵌合类器官。然而,上述所有 scCRISPR-seq 平台都仅限于体外应用。相比之下,体内测定更具吸引力,因为它们与真实的有机条件更相似。因此开发了体内 Perturb-seq,这是 Perturb-seq 方案的一种变体,涉及在体内进行的合并扰动。此外,PoKI-seq (Roth et., 2020) 已经证明了体内研究重编程 T 细胞对实体瘤的免疫反应的可行性。最近,Santinha 等人。(2023) 开发了腺相关病毒 (AAV) 介导的直接体内单细胞 CRISPR 筛选,称为 AAV-Perturb-seq,一种可用于体内遗传扰动转录连锁分析和表型分析的可调且广泛适用的方法。
基于表观基因组的 scCRISPR-seq
基于表观基因组的 scCRISPR-seq 除了转录组应用外,还有基于表观遗传学的 scCRISPR-seq 平台。2019 年开发了 Perturb-ATAC,这是一种将 CRISPR 干扰或敲除与单细胞染色质可及性分析相结合的方法,该方法基于通过测序检测转座酶可及性染色质 (ATAC-seq) 同时检测 CRISPR 向导 RNA 和开放染色质位点。他们应用这种方法来确定各种反式调控因子的作用,包括 TFs、染色质修饰剂以及人类和病毒 ncRNA,这可能有助于剖析顺式调控元件和 ncRNA 转录本已被证明对基因表达有影响的基因座(Cho 等人,2018b;Engreitz 等人,2016 年;Rubin 等人,2019 年)。Perturb-ATAC 将 scCRISPR-seq 研究扩展到表观基因组领域,使 scCRISPR-seq 更强大、应用更广泛。然而,Perturb-ATAC 受到高成本和低吞吐量的限制。为了应对这一限制,开发了 Spear-ATAC(Pierce 等人,2021 年)以实现显着更高的细胞通量和大幅降低成本,提供了更实用的替代方案。此外,CRISPR-sciATAC (Liscovitch-Braueret al., 2021) 显示出与 Spear-ATAC 相似的细胞通量和成本。然而,它对染色质可及性的细微变化表现出有限的敏感性。
多模态 scCRISPR-seq
多模式单细胞检测提供了异质细胞群的高分辨率快照,但上述 scCRISPR-seq 平台都仅限于一种模式,例如转录组或表观基因组。因此,为了将该技术同时应用于多组学,开发了多模态 scCRISPR-seq。Xie et al. (2017) 通过索引 CRISPR 测序 (Mosaic-seq) 开发了Mosaic单细胞分析,以扰乱增强子并共同测量每个细胞的转录组及其诱导的 sgRNA。Mosaic-seq 提供了一种新颖的工具,以基于 inper扰动的方式询问非编码基因的功能。此外,Mimitou 等人。(2019) 通过测序开发了扩展的 CRISPR 兼容转录组和表位细胞索引 (ECCITE-seq),它允许同时检测来自每个细胞的转录组、蛋白质、克隆型和 CRISPR 扰动。通过构建 ECCITEseq 抗体的 a49 标记面板来分析人外周血单核细胞 (PBMC),他们恢复了许多重要结果(Fanok等人,2018 年;Stoeckius 等人,2017 年),证明了 ECCITE-seq 结合免疫表型、克隆型和转录组信息的能力。空间转录组学能够表征基因表达谱,同时保留有关空间组织背景的信息,这为生物学的不同领域提供了新的见解,例如神经科学、发育生物学和癌症研究(Moses 和 Pachter,2022)(参见下面的空间部分)。最近,开发了一种新的多模态 scCRISPR-seq,称为 Perturb-map (Dhainaut et., 2022),以通过成像和空间转录组学实现 CRISPR 筛选原位的多模态表型分析。Perturb-map 基于蛋白质条形码 (Pro-Code) 系统,该系统使用几个线性表位的三重体组合来创建更高阶的唯一条形码集(Wroblewska 等人,2018 年)。这些独特的条形码可以标记表达不同 CRISPR gRNA 的细胞。需要注意的是,Perturb-map 是唯一能够实现体内 CRISPR 筛选与空间转录组相结合的 scCRISPR-seq 平台,特别适用于鉴定肿瘤组成、组织和免疫的遗传决定因素。Perturb-map 基于蛋白质条形码 (Pro-Code) 系统,该系统使用几个线性表位的三重体组合来创建更高阶的唯一条形码集(Wroblewska 等人,2018 年)。这些独特的条形码可以标记表达不同 CRISPR gRNA 的细胞。需要注意的是,Perturb-map 是唯一能够实现体内 CRISPR 筛选与空间转录组相结合的 scCRISPR-seq 平台,特别适用于鉴定肿瘤组成、组织和免疫的遗传决定因素。Dhainaut 等人。(2022) 将 Perturb-map 应用于 TME 的研究。他们在肺癌小鼠模型中敲除 35 个基因,发现敲除 Tgfbr2 可以促进 TME 重塑和免疫排斥。
scCRISPR-seq 数据分析工具
scCRISPR-seq 数据包含丰富的扰动信息,这对于在单细胞水平上探索基因型和表型之间的关联具有天然的优势。例如,通过将 Perturb-seq 应用于 K562 细胞系,Adamson 等人。(2016) 表明,PERK 的扰动对未折叠蛋白质反应的影响比 ATF6 和 IRE1α 更大。Datlinger 等人 (2017) 在用 CROP-seq 激活 T 细胞受体 (TCR) 的条件下扰动了 Jurkat 细胞系中的 23 个转录因子,发现 LCK 、 ZAP70 和 LAT 的敲除对 TCR 激活信号传导有很强的负面影响。然而,由于其固有的噪声,scCRISPR-seq 数据的分析是一项重大挑战。因此,已经开发了几种生物信息学工具来帮助分析 scCRISPR-seq 数据(支持信息中的表 S12)。通常,这些 scCRISPR-seq 数据分析工具侧重于三个部分(图 16B):(i) 数据预处理,包括质量控制、归一化和差异表达基因检测,例如 MIMOSCA 、MUSIC和 SCREE 。(ii) 数据去噪,包括单细胞插补、转义细胞过滤和复合因子分解,例如 MUSIC、mixscape 和 SCREE 。(iii) 功能分析,包括确定每个扰动影响的优先级,确定每个扰动的功能,推断调节网络和基因相互作用,例如 MUSIC、Normalisr、scMAGeCK、Pando和 GEARS。具体来说,LRICA是通过低秩矩阵分解来解码数据的驱动信号/分量。MIMOSCA是一种计算sg RNA与每个基因之间关系的计算框架。LRICA 和 MIMOSCA 是作为原型开发的,没有可执行且用户友好的实现。因此,Duan 等人。(2019) 开发了 MUSIC,这是一个通用的计算框架,用于通过主题建模评估每个扰动的影响,它最初出现在机器学习和自然语言处理社区或特定文档集中的潜在主题发现中。MUSIC 将基因型表型与对大量噪声的耐受性联系起来,并从三个角度分析 scCRISPR-seq 数据,即优先考虑基因扰动效应作为整体扰动效应、非功能性主题特异性方式,以及量化不同扰动之间的相关性。scMAGeCK 也是分析 scCRISPR-seq 数据的框架,它是从 MAGeCK 扩展而来的。scMAGeCK 包括两个模块,scMAGeCK-RRA 和 scMAGECK-LR,其中 scMAGeCK-RRA 用于通过负二项分布识别显著富集的 sgRNA,scMAGeCK-LR 用于通过线性回归评估受影响的基因。scMAGeCK 显示出对假阳性的良好控制,并且比其他方法具有更好的灵敏度。除了 scCRISPR-seq 的通用计算框架外,一些工具还专注于数据去噪。例如,SCEPTRE 是为使用条件随机化测试进行 scCRISPR-seq 数据校准而开发的。SCEPTRE 对 scCRISPR-seq 数据表现出良好的校准和灵敏度,产生了数百种由正交生物学证据支持的新调控关系。Mixscape 旨在通过混合判别分析过滤逃逸的细胞 (细胞诱导的 sgRNA,但不表现出扰动效应) 来提高 scCRISPR-seq 数据的信噪比。Normalisr 用于重建 scCRISPR-seq 数据的基因调控网络。Wang 等人.(2022g) 强调了识别克隆细胞的重要性,因为它们可能导致 scCRISPR-seq 数据出现假阳性。SCREE 是 scCRISPR-seq 数据分析的综合管道。与前面提到的最初专注于 scCRISPR-seq 中的数据去噪和挖掘的方法相比,GEARS 专门设计用于预测对单基因和多基因扰动的转录反应。这些方法大大增强了 scCRISPR-seq 数据的分析。
scCRISPR-seq 的应用
scCRISPR-seq 因其强大的能力而被广泛应用于各个领域,包括连接基因型与表型、剖析遗传调控以及研究肿瘤和自闭症等非特异性疾病的遗传机制。
连接基因型和表型
与传统的 CRISPR 筛选只能识别具有非常不同表型的基因相比,scCRISPR-seq 具有揭示任何基因功能的能力。因此,scCRISPR-seq 天然适合大规模地将基因型与表型联系起来。例如,Jaitin 等。(2016) 揭示了 22 个 TFs 对 CRISP-seq 刺激的脂多糖 (LPS) 刺激的出生骨髓细胞 (BMC) 的抗病毒、炎症或发育过程的调节作用。Adamson 等人。(2016) 通过 Perturb-seq 系统分析了 K562 细胞中 83 个未折叠蛋白反应 (UPR) 相关基因的影响。此外,基因组规模的 Perturb-seq(Replogle等人,2022 年)提供了对遗传扰动(9,867 个基因)的公正、全面的分析,有助于系统剖析与基因翻译和核糖体生物发生相关的基因之间的关系。
剖析遗传调控
scCRISPR-seq 还用于剖析基因组元件之间的复杂关系,包括编码基因、转录因子、染色质调节因子、增强子和其他非编码元件。例如,Adamson 等人。(2016) 使用 Perturb-seq 发现了三个 UPR 传感器基因(ATF6、PERK 和 IRE1)之间的串扰。CROP-seq 扰乱了 LPS 刺激后 Jurkat 细胞中调节 TCR 激活的 TFs,并揭示了 TFs 之间的关系。此外,用于增强子扰动的 scCRISPR-seq,例如 Mosaic-seq (Xie et tal., 2017),可以发现新的增强子-基因对。此外,scCRISPR-seq 与 scATAC-seq 偶联,如 Perturb-ATAC、Spear-ATAC 和 CRISPR-sciATAC 可以揭示人淋巴细胞和白血病细胞中的表观遗传景观重塑剂
研究遗传机制
有几种体内 scCRISPR-seq 平台可用,能够研究肿瘤和自闭症等非特异性疾病的遗传机制。例如,Perturb-map (Dhainaut et., 2022) 有助于识别与肿瘤组成、组织和免疫相关的遗传决定因素。Using Perturbmap, Dhainaut etal.(2022) 发现 TGFBR2 肺癌细胞的敲除促进了肿瘤微环境重塑和免疫排斥。Roth 等人。(2020) 使用 PoKI-seq 对增强 T 细胞抗肿瘤功能的嵌合抗原受体进行了筛选,提高了免疫抑制条件下的肿瘤浸润和细胞杀伤率 内黑色素瘤。此外,Jin 等人。(2020b) 使用 invivo Perturb-seq 评估了 35 个与自闭症谱系障碍/神经发育迟缓 (ASD/ND) 相关的 denovo 功能丧失风险基因。他们从神经元和神经胶质细胞类别中鉴定了细胞类型特异性和进化上保守的基因模块。利塔尔。(2023) 还关注了这些高危自闭症谱系障碍基因,他们揭示了它们对使用 CHOOSE 系统在嵌花类器官中决定细胞命运的影响。最近,Santinha 等人。(2023) 使用 AAV-Perturb-seq 系统分析了与成年小鼠大脑前额叶皮层中 22q11.2 缺失综合征基因相关的表型景观。他们确定了三个 22q11.2 连锁基因,这些基因积极参与已建立的和以前未识别的体内神经元功能控制途径。
总结
在本章中,我们对 scCRISPR-seq 进行了全面的回顾,分为三个不同的部分,其中包括 scCRISPR-seq 的类别、scCRISPR-seq 数据分析的工具以及 scCRISPR-seq 的显着应用。scCRISPR-seq 一直是功能基因组学研究的强大方法 (Bock et al., 2022)。在本节中,我们根据其综合组学方法将 scCRISPR-seq 分为三个主要类别:基于转录组的 scCRISPRseq、基于表观基因组的 scCRISPR-seq 和多模态 scCRISPRseq。鉴于 scCRISPR-seq 数据中固有的噪声,已经开发了多种生物信息学工具来帮助初始化分析,从而没有显着改进。
scCRISPR-seq 的多功能性使其在各个领域得到广泛应用,提供了强大的功能,例如连接基因型与表型、剖析遗传调控以及探索肿瘤和自闭症等特定疾病的遗传机制。然而,在生物学研究中更广泛地采用之前,需要注意三个关键方面:(i) 降低复杂性和成本:应努力进一步简化和降低 scCRISPR-seq 实验的复杂性和成本。这将增强可扩展性和可及性,使更多实验室能够利用这项技术。(ii) 扩大对复杂组织和体内设置的适用性:虽然目前的 scCRISPR-seq 平台主要针对细胞系,但迫切需要开发更强大的 scCRISPR-seq 平台,这些平台可以应用于更复杂的组织,包括类器官,以及理想的体内设置。这种扩展将使更广泛的生物学研究成为可能。(iii) 降噪技术:随着 scCRISPR-seq 平台数量的增加,开发更强大的方法来破译 scCRISPR-seq 数据中的固有噪声变得至关重要。这些方法将有助于 scCRISPR-seq 结果的可靠性和可解释性,进一步提高它们在不同研究环境中的实用性。
结尾
scRNA-seq 技术引起了世界各地许多科学家的广泛关注,因为它具有在单细胞水平上研究细胞异质性的优势。自新时代 inscRNA-seq 研究成立以来,已经过去了仅仅 14 年,在此之前,Tang 等人取得了初步的概念和技术突破。(2009) 在 2009 年。在测序技术和生物信息学不断发展的推动下,scRNA-seq 研究领域目前正在经历研究的激增。scRNA-seq 技术的成熟极大地促进了其他单细胞组学研究的进步。目前,单细胞组学检测已扩展到基因组 (Dey et al., 2015)、表观基因组 (Muto et al., 2021)、空间转录组学 (Chen et al., 2022)、蛋白质组学 (Petersonet al., 2017;Specht et al., 2021)、代谢组学 (Shrestha, 2020) 等多组学水平 (Angermueller et al., 2016),为单细胞水平研究提供更全面、更精细、更完整的分析策略。在这篇综述中,我们总结了单细胞组学技术、数据分析及其应用的最新进展,概述了单细胞测序领域跨多个层次的前景。
在第 1 章中,我们全面概述了目前可用的 scRNA-seq 技术、实验方法、数据分析程序及其在生物医学领域的应用。最初,通过分离单细胞并独立构建测序文库来进行单细胞测序。这些单细胞测序技术只能检测少量细胞(数到数百个),例如 Tang 方法、STRT-seq 和 SMART-seq(Islam 等人,2012 年;Ramsköld 等人,2012 年;Tang 等人,2009 年)。然而,随着对测序技术的深入研究,基于条形码标签的单细胞鉴定已经出现,以及基于微滴或微孔的新型单细胞分离技术的出现,如 Drop-Seq 和 Cyto-Seq (Fan et., 2015a;Macosko 等人,2015 年),单细胞转录组测序已进入高通量时代。测序成本显著降低,同时自动化和通量显著提高。ScRNAseq 技术解决了细胞异质性问题,为临床疾病尤其是肿瘤的个体化治疗开辟了新途径,促进了精准医疗的发展。然而,由于起始材料量少,scRNA-seq 存在 oflow 捕获效率和高脱落率的局限性。与大量 RNA-seq 相比,scRNA-seq 产生的数据噪声更大且可变性更强。尽管研究人员已经设计了多种工具来进行不同的 scRNA-seq 数据分析,但技术噪声和生物变异(例如,随机转录)仍然对 scRNA-seq 数据的计算分析构成巨大挑战(Chen 等人,2019a)。因此,数据分析方法仍需进一步优化和改进。
与日益成熟的 scRNA-seq 技术相比,其他单细胞组学技术仍处于萌芽阶段。在第 2、3、4 和 5 章中,我们重点介绍了过去十年中单细胞基因组、表观基因组、蛋白质组学和代谢组学测序的最新工具、计算方法和应用。ScWGS 彻底改变了我们对遗传变异及其对人类健康和疾病影响的理解。它的快速发展加速了基因组研究,实现了个性化医疗,并为疾病的遗传基础和人类基因组多样性提供了有价值的见解。细胞在染色质可及性、核小体定位、组蛋白修饰和 DNA 甲基化方面表现出广泛的异质性。在单细胞样品中绘制这些表观基因组信息对于发育生物学、癌症研究以及很快的发展非常重要。单细胞表观基因组测序方法的进步使单细胞染色质状态的高分辨率图谱成为可能。然而,如今,单细胞表观基因组技术存在数据丢失的问题。因此,尽管单个细胞表观基因组数据集是聚类分析和基于大量目标位点集合揭示细胞异质性的强大资源,但它们提供单个目标位点信息的能力非常有限(Carter 和 Zhao,2021)。因此,在未来的研究中,需要提高各种单个细胞表观基因组测定中染色质靶位点的覆盖率,这将有助于理解整个细胞水平和单个特异性位点的细胞异质性。单细胞蛋白质组学由于其成分复杂、丰度低、动态范围宽、缺乏扩增能力,处于爆发性发展的早期阶段。就在 2019 年,对单细胞蛋白质组的分析被描述为“梦想”,但今天已经开发了几种有前途的工具(Marx,2019)。我们相信,随着可及性的优化和通量的进一步提高,单细胞蛋白质组学在科学和临床研究中真正的大规模应用,如器官图谱、药物筛选和精确的疾病分类,是触手可及的。单细胞代谢组学用于鉴定单细胞中代谢物的组成,测量其丰度,并研究其动态变化。同时,代谢组代表基因组、转录组和蛋白质组的下游产物,并提供了功能的更直接和动态快照(Shrestha,2020 年)。总体而言,单细胞组学技术仍处于萌芽阶段,它们将继续蓬勃发展。
单个细胞是生命的基本单位。对单个细胞进行多组学分析可以深入了解细胞的表型、疾病状态和环境影响。在第 6 章、第 7 章和第 8 章中,我们全面总结了多组学的综合分析、scRNA-seq 和 CRISPR 筛选的联合应用以及空间转录组。在复杂的生物过程中,例如肿瘤发生和衰老,异质性发生在不同的层面,包括基因组、转录组、蛋白质组和表观基因组。如果仅从单个细胞 atatime 分析一个成分,则只能检测到基因调控网络的局部概况,而无法准确预测复杂的全球情况。在这种情况下,多组学技术凸显其独特的优势,可以在复杂组织的研究中提供更完整的基因调控网络图谱。对于空间转录组学,它能够在保留空间信息的情况下测量基因表达,这将有利于研究细胞间关系和发现空间背景下的新调控机制。此外,空间转录组学使探索细胞命运决定的空间调控机制和组织模式的结构成为可能。与传统的 CRISPR 杂交筛选相比,scRNA-seq 和 CRISPR 的组合不仅可以在单个实验中筛选数千个 gRNA,还可以同时捕获扰动的全转录组数据,以最清楚地了解细胞类型特异性基因功能和通路分析。因此,这些技术的结合可以更好、更深入地理解关键的生物过程和机制,这是未来单细胞技术发展的重要方向。
如今,单细胞组学技术在通量和分辨率方面都取得了重大进步。展望未来,单细胞技术发展的主要趋势是提高单细胞分选的效率和通量,增强测序覆盖率和灵敏度,实现高通量多组学研究,并开发更多自动化的单细胞技术平台,这将有助于降低单细胞技术的成本和技术门槛。单细胞技术有望在科学研究和研究转化领域得到广泛应用,并将对健康监测、疾病诊断和治疗做出巨大贡献。
参考文献:
Sun F, Li H, Sun D, et al. Single-cell omics: experimental workflow, data analyses and applications. Sci China Life Sci. 2025;68(1):5-102. doi:10.1007/s11427-023-2561-0